S/W VASP
Precision Computational Modeling for Material Science and Engineering
VASP (Vienna Ab initio Simulation Package) is a leading software suite for quantum mechanical simulations of materials. Built on the foundation of density functional theory (DFT), Hartree-Fock approximations, and advanced hybrid methods, VASP offers unparalleled accuracy and efficiency for solving complex material-related problems. Its state-of-the-art algorithms are ideal for research and industrial applications requiring predictive modeling at the atomic scale.
VASP’s advanced methods include:
- DFT with both local and hybrid functionals for high accuracy.
- Inclusion of van der Waals interactions for studying weakly bonded systems.
- GW calculations and time-dependent DFT for electronic excitations.
- Efficient algorithms for large-scale systems and parallel computing.
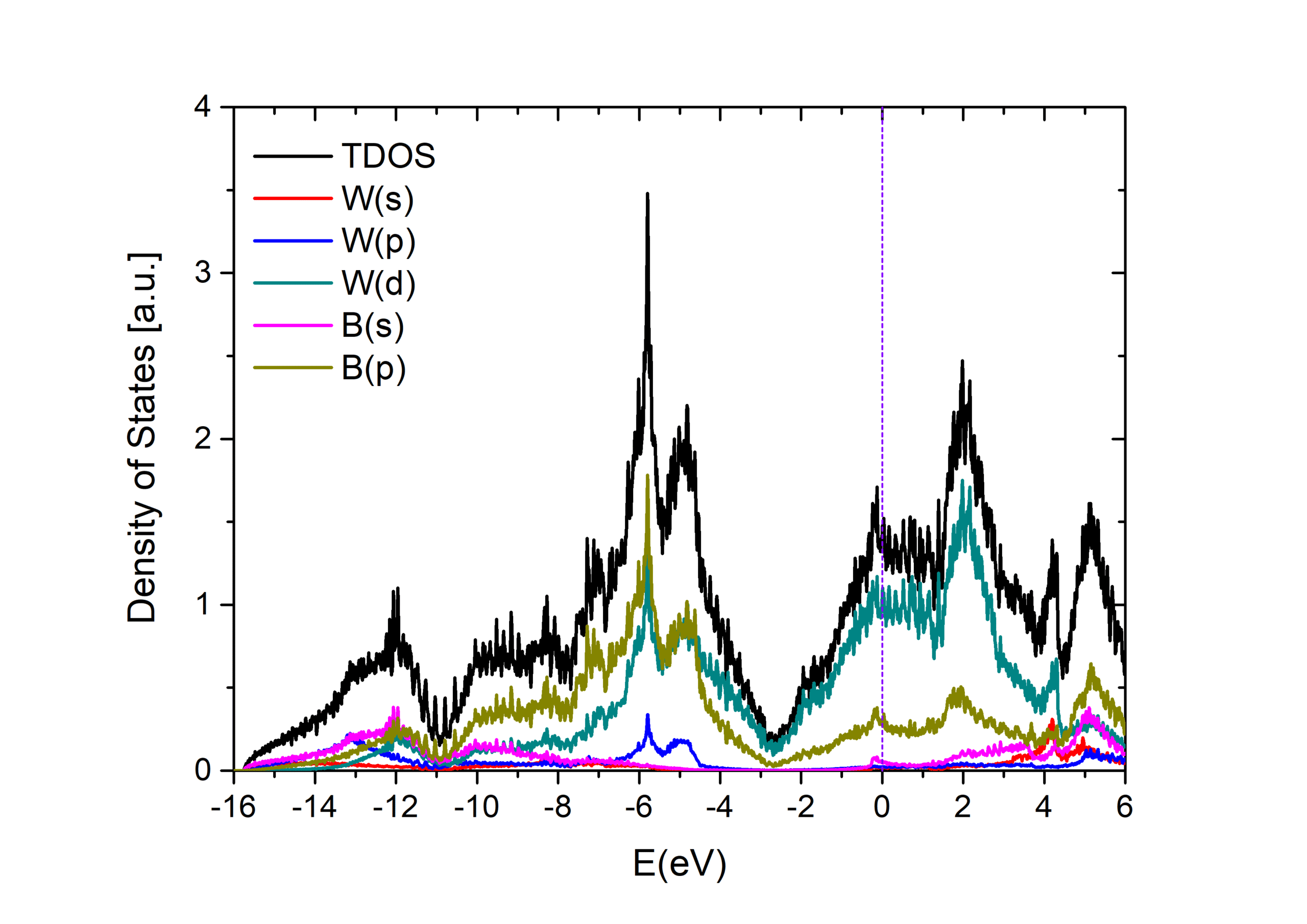
Our expertise
Our team specializes in leveraging VASP to deliver cutting-edge computational solutions tailored to research or industrial needs providing comprehensive modeling services in the following areas:
- Electronic Structure Calculations
- Calculation of band structures, density of states (DOS), and Fermi surfaces.
- Precise determination of electronic properties for semiconductors, insulators, and metals.
- Insights into charge transfer and electronic density mapping.
- Structural Optimization
- Energy minimization to determine ground-state atomic arrangements.
- Prediction of lattice parameters, bond lengths, and angles.
- Vibrational and Thermodynamic Properties
- Phonon dispersion and density of states for understanding vibrational modes.
- Thermodynamic properties such as free energy, entropy, and heat capacity.
- Analysis of thermal conductivity and stability across temperature ranges.
- Ab Initio Molecular Dynamics (AIMD)
- Simulation of dynamic behavior at finite temperatures.
- Exploration of phase transitions, diffusion processes, and thermal effects.
- Real-time tracking of atomic and molecular motion.
- Advanced Material Properties
- Magnetism: Spin-polarized calculations, magnetic moments, and spin-orbit coupling.
- Optical Properties: Dielectric constants, refractive indices, and absorption spectra.
- Defect Chemistry: Formation energies and electronic effects of vacancies and impurities.
- Materials Design and Discovery
- Prediction of properties for novel materials, including 2D materials, alloys, and composites.
- Exploration of energy materials such as catalysts, batteries, and photovoltaics.
- Surface and interface studies for adsorption, catalytic activity, and electronic behavior.
Proven Expertise: Years of experience in theoretical modeling and computational physics.
Cutting-edge Resources: Access to powerful computing infrastructure for large-scale simulations.
Custom Solutions: Fully adaptable workflows tailored to your specific material challenges.
Collaborative Approach: Support for both short-term projects and long-term research partnerships.
Let’s Collaborate
Harness the full potential of VASP for your materials research or development project. Contact us to explore how we can support your work with precise, reliable, and high-performance computational solutions.
